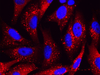
Los investigadores han utilizado el sistema CRISPR de edición del genoma para determinar qué variantes genéticas presentes en pacientes con síntomas compatibles a la deficiencia de CAD comprometen la función de la proteína. A partir de esta información han detectado 11 niños candidatos para ser tratados con un suplemento nutricional que mejora los síntomas y que posiblemente podrán beneficiarse del tratamiento con uridina. “La administración de uridina tiene un efecto terapéutico espectacular”, ha señalado Hudson Freeze, investigador del Sanford Burnham Prebys Medical Discovery Institute y uno de los directores del trabajo. “Con este ensayo, podemos proporcionar esperanza a algunas familias, al tiempo que salvamos a otros de expectativas poco realistas, lo que es increíblemente importante «. También ha participado en el estudio el equipo de Santiago Ramón-Maiques, científico titular e investigador CIBERER del Instituto de Biomedicina de Valencia.
La deficiencia de CAD es una enfermedad metabólica tan poco frecuente que hasta el momento solo existen 17 personas diagnosticadas en el mundo. Está causada por cambios en el gen CAD que afectan a la función de la proteína que codifica, una proteína citoplásmica que interviene en una ruta metabólica esencial para el organismo.
Estudios recientes han encontrado que el tratamiento con uridina, un suplemento nutricional, mejora significativamente los síntomas de la deficiencia en CAD y retrasa su progreso. Sin embargo, el carácter no específico con el que se manifiesta la enfermedad, que comparte síntomas con otras condiciones, dificulta poder decidir qué pacientes se beneficiarán de la terapia con uridina. Además, el gen CAD es muy grande y, aunque se han descrito más de 1000 cambios que afectan a la secuencia de aminoácidos de la proteína, se desconoce exactamente cuáles de ellos comprometen la función de la proteína.
Ante la posibilidad de un tratamiento terapéutico tan sencillo como un suplemento nutricional, los investigadores se plantearon desarrollar un método para identificar qué pacientes tienen una probabilidad elevada de responder de forma positiva y recurrieron a la herramienta CRISPR para hacerlo posible.
En primer lugar, los investigadores identificaron a 25 pacientes que podrían ser candidatos a la terapia con uridina. Todos ellos tenían síntomas similares a los derivados de la ausencia de función de CAD y eran portadores de variantes genéticas en ambas copias del gen CAD.
A continuación, el equipo utilizó CRISPR para inactivar el gen CAD y desarrollar una línea de células humanas deficitarias para la proteína CAD. Por sí mismas estas células no pueden sobrevivir. Pero al igual que ocurre con los pacientes con déficit de CAD, el tratamiento con uridina recupera la funcionalidad de las células y favorece su supervivencia.
El siguiente paso de los investigadores fue evaluar lo que ocurre en las células deficitarias de CAD cuando se incorpora cada una de las variantes del gen identificadas en los pacientes candidatos. Si la adición de la variante rescata la supervivencia de las células esto significa que la variante no compromete completamente la función de la proteína. Si por el contrario, no lo hace, será debido a que la variante evita que se produzca proteína CAD funcional.
A través del ensayo celular los investigadores han detectado que 11 de los 25 pacientes evaluados tienen ambas copias del gen CAD dañadas y son firmes candidatos a ser tratados con un suplemento nutricional de uridina.
Los resultados del trabajo tienen consecuencias inmediatas para los 11 niños pero también resultan relevantes para otros pacientes. “Hemos desarrollado una prueba rápida y fiable sobre los efectos de muchas de las variaciones en el gen CAD sobre la función de la proteína producida a partir de este gen», explica Santiago Ramón-Maiques. «Dicha información es muy importante para decidir si seguir buscando en otros genes mutaciones causales de los síntomas o si comenzar inmediatamente la terapia con uridina».
Las enfermedades raras como la deficiencia de CAD son tan poco frecuentes que resulta complicado identificar suficientes pacientes como para estudiar los mecanismos que intervienen en ellas, así como encontrar tratamientos que beneficien a los pacientes. El tratamiento con uridina resulta beneficioso para los pacientes que carecen de proteína CAD funcional, lo que representa una oportunidad para los pacientes y sus familias.
«CAD es realmente una máquina increíblemente complicada y hemos dedicado los últimos 10 años a entender cómo funciona», destaca Ramón-Maiques. «Este ensayo nos permitirá identificar las variantes dañinas y ayudar a los niños afectados a obtener el mejor tratamiento posible. Este trabajo demuestra cómo los estudios básicos, sin una finalidad principalmente curativa, pueden tener increíbles efectos en el tratamiento de pacientes afectados”.
Referencia: del Caño-Ochoa, F., Ng, B.G., Abedalthagafi, M. et al. Cell-based analysis of CAD variants identifies individuals likely to benefit from uridine therapy. Genet Med (2020). https://doi.org/10.1038/s41436-020-0833-2
Fuente: Un nuevo test para una enfermedad metabólica identifica niños que pueden beneficiarse de un tratamiento sencillo. https://www.ciberer.es/noticias/un-nuevo-test-para-una-enfermedad-metabolica-identifica-ninos-que-pueden-beneficiarse-de-un-tratamiento-sencillo

AtCDF3 gene induced greater production of sugars a...
Un estudio con datos de los últimos 35 años, ind...
Un equipo de investigadores de la Universidad Juli...

En nuestro post hablamos sobre este interesante tipo de célula del...

Horizon ha puesto en funcionamiento una nueva planta dedicada íntegra...