Las enfermedades del neurodesarrollo tienden a recibir una menor atención que otras enfermedades que afectan al sistema nervioso, pero aun así siguen suponiendo un importante reto para la medicina actual, debido a su complejidad en cuanto a factores causantes y sintomatología derivada. Estos trastornos son debidos a cualquier tipo de fallo que pueda producirse durante el proceso de desarrollo y maduración del sistema nervioso neonato. Entre las múltiples causas asociadas, se conocen cientos de mutaciones en genes implicados principalmente en la remodelación de la cromatina, la función sináptica o la regulación de la transcripción de otros genes.
Un gen que merece especial atención es el gen MECP2. Localizado en el cromosoma X, este gen permite la expresión de la proteína MeCP2, un regulador de la transcripción de numerosos genes que ha sido relacionado con los procesos de diferenciación y maduración neuronal. Debido a su gran distribución y amplio rango de acción, las alteraciones del gen MECP2 desencadenan una patología diversa a la par que compleja. Un ejemplo de ello es el síndrome de Rett, una enfermedad poco frecuente que afecta a 1 de cada 10.000 niñas nacidas en todo el mundo. En el 95% de los casos, este síndrome es debido a mutaciones directas en el gen MECP2, mientras que el 5% restante son consecuencia de alteraciones en las regiones que regulan la expresión del mismo (Zoghbi et al., 1999).
La relevancia clínica del síndrome de Rett radica en su amplia sintomatología, que incluye desde la pérdida progresiva de habilidades motoras y del habla, hasta el desarrollo de problemas respiratorios, crisis epilépticas y discapacidad intelectual. También comparte algunas características con el autismo, como son los movimientos repetitivos de manos y las alteraciones sociales (Hagberg, 2002). Todo ello deriva en una disminución tanto de la calidad como esperanza de vida de las niñas afectadas. Además, al tratarse de un gen localizado en el cromosoma X, las consecuencias son mucho más graves en los niños, que no suelen sobrevivir a su primer año de vida.
Durante muchos años, las alteraciones sociales de las niñas con Rett han sido la principal causa de que fuese considerado como un trastorno de espectro autista (TEA). Sin embargo, con el tiempo se ha visto que estas niñas no huyen del contacto social, sino que incluso llegan a buscarlo con mayor frecuencia. Por este motivo, consideramos necesario investigar en mayor profundidad las bases neurobiológicas que propician las alteraciones sociales en Rett.
Se ha demostrado que algunos nonapéptidos como la oxitocina y la vasopresina desempeñan un papel fundamental en la modulación de comportamientos sociales tanto en humanos como otros mamíferos. Además, algunos estudios han encontrado desregulaciones en el sistema nonapeptidérgico de modelos animales que se emplean para estudiar diversos trastornos del neurodesarrollo. Por este motivo, algunos de estos nonapéptidos, como la oxitocina, se vienen proponiendo desde hace años como potenciales dianas terapéuticas sobre las que actuar en casos de TEA. Decidimos, por tanto, realizar un estudio inmunohistoquímico que nos permitiese analizar de forma simultánea los sistemas de inervación tanto de vasopresina como de oxitocina en un modelo de ratón de síndrome de Rett que ha sido previamente validado (Guy, Hendrich, Holmes, Martin y Bird, 2001).
Resulta curioso que, a pesar de tratarse de un síndrome que afecta fundamentalmente a niñas, gran parte de la bibliografía científica referente al síndrome de Rett está basada en estudios realizados con ratones machos. Por este motivo, para nuestro estudio trabajamos tanto con ratones macho, que son nulos para el gen Mepc2 y reflejan mejor los efectos de la ausencia total de proteína MeCP2, como con hembras heterocigotas para la mutación, cuyas condiciones fenotípicas se asemejan más a las de las niñas afectadas.
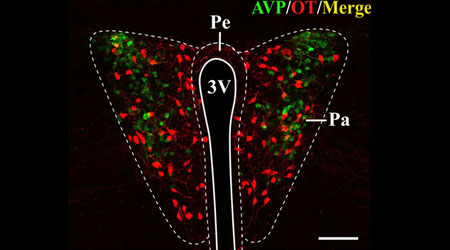
En lo relativo al sistema vasopresinérgico, la inervación de algunos núcleos del llamado “cerebro social” es sexualmente dimórfica, y su abundancia depende de los niveles circulantes de testosterona. Esto implica que haya inervación en determinadas regiones del cerebro de ratones macho, que se encuentra ausente en el cerebro de las hembras (Otero-Garcia et al., 2014; Rood et al., 2013). Al comparar ratones nulos para el gen Mecp2 con ratones sanos, encontramos una disminución bastante notable de la inervación vasopresinérgica en estos primeros, llegando a encontrarse incluso ausente en algunos de estos nodos del cerebro social. Este déficit podría deberse a una posible regulación directa de la proteína MeCP2 sobre la expresión de vasopresina. Por otro lado, tras cuantificar y analizar el número de neuronas productoras de oxitocina y vasopresina, así como la mayor parte de inervación vasopresinérgica y oxitocinérgica que no es dependiente de testosterona en el cerebro de ratones, no encontramos diferencias que pudieran deberse a la falta de la proteína MeCP2, ni en machos ni en hembras. Esto apunta a que las mutaciones en el gen Mecp2 afectan al desarrollo cerebral de manera indirecta, a través de alteraciones en el desarrollo sexual. De hecho, los machos mutantes para el gen Mecp2 presentan testículos internos, reforzando esta hipótesis. Otra posible explicación para la ausencia de diferencias entre genotipos en el caso de las hembras estaría relacionada con la edad de los ratones de nuestro estudio, ya que mientras los ratones nulos para el gen Mecp2 son plenamente sintomáticos y su esperanza de vida no se alarga mucho más, las hembras heterocigotas no comienzan a manifestar la sintomatología hasta los 3-6 meses de vida, y en nuestro estudio analizamos animales de 2 meses.
A la vista de todos estos datos, decidimos analizar los niveles de testosterona en nuestros machos mutantes. Para ello, llevamos a cabo una medida indirecta, midiendo en su orina los niveles de determinadas feromonas sexuales, como la darcina, cuya expresión está demostrada que es dependiente de los niveles de testosterona. De acuerdo con nuestra hipótesis, la darcina desaparece de la orina de machos mutantes, y la producción de otras feromonas se encuentra reducida en comparación con los machos sanos.
Para finalizar, analizamos algunos de los comportamientos típicos de ratones machos que son regulados por la testosterona, y a los que contribuye la inervación vasopresinérgica sexualmente dimórfica. Entre estos comportamientos se incluyen algunos como la agresión hacia otros machos y el grado de sociabilidad con sus congéneres. Para analizar la agresividad, realizamos un test conocido como “resident-intruder” en el que se coloca un ratón intruso en la jaula donde se encuentra el ratón que queremos estudiar. Como resultado, observamos que los ratones Mecp2-nulos no atacan nunca al intruso, cosa que suele ser común en ratones sanos. Además, tienden a investigar menos al ratón intruso, y a emplear más tiempo acicalándose, algo que se ha asimilado con comportamientos repetitivos en humanos.
Son varias las controversias con respecto a si los ratones Mecp2-nulos son más o menos sociables que los ratones normales. Mientras que algunos estudios realizados con otros modelos de ratón de Rett han detectado que los ratones mutantes suelen menos sociables, otros defienden justo lo contrario. Para poder analizar si un animal es más o menos sociable, se le suele dar a elegir entre pasar tiempo cerca de un cilindro vacío o de otro que contiene a un congénere. Al evaluar cómo de sociables eran nuestros ratones Mecp2-nulos, observamos una mayor preferencia por parte de estos a pasar más tiempo cerca de su congénere. Aunque a primera vista, estos resultados parecen contradecirse con los obtenidos en el test “resident-intruder”, donde los Mecp2-nulos tienden a investigar menos al intruso, es importante tener en cuenta que en el resident-intruder los ratones se encuentran en estrecho contacto dentro de la misma caja, mientras que en este segundo test se encuentran separados, por lo que no existe intimidación por ser los Mecp2-nulos de menor tamaño que los ratones sanos. Además, los ratones nulos parecen igual de capaces que los sanos a la hora de reconocer a otros congéneres. Estos comportamientos están asociados a la inervación oxitocinérgica, la cual no parece estar afectada en los ratones Mecp2.
En resumen, los ratones con mutaciones en el gen Mecp2, carecen de la inervación vasopresinérgica que forma parte del cerebro social y que es dependiente de los niveles circulantes de testosterona. Además, estos machos no producen feromonas sexuales, que también son dependientes de testosterona, tampoco son agresivos y tienden a ser más sociables.
Como conclusión, nuestros resultados no parecen apoyar que la vasopresina u oxitocina sean potenciales dianas terapéuticas para las niñas afectadas de Rett, puesto que no hemos encontrado déficits en las hembras. No obstante, sería necesario trabajar también con hembras sintomáticas, de al menos 6 meses de edad, para poder descartar definitivamente que no existen déficits. No obstante, nuestros resultados resaltan que, al estudiar un síndrome del neurodesarrollo, y concretamente aquellos que cursan con alteraciones del comportamiento social, es importante tener en cuenta también los efectos de estas mutaciones sobre el desarrollo sexual. Estos resultados pueden ser de ayuda para entender otras condiciones relacionadas con el gen Mecp2, como es el caso del síndrome de duplicación del gen Mecp2 que afecta principalmente a niños, los cuales parece que tienden a ser más agresivos (Tantra et al., 2014). Además, otros modelos de ratón de síndromes del neurodesarrollo, como es el caso del síndrome de X Frágil, también prevalente en niños, muestran de igual modo fenotipos testiculares alterados. Sería pues, muy interesante poder estudiar también los sistemas nonapeptidérgicos en dichos modelos.
Artículo original: Martínez-Rodríguez E, et al. Male-specific features are reduced in Mecp2-null mice: analyses of vasopressinergic innervation, pheromone production and social behaviour. Brain Structure and Function. 2020. DOI: https://doi.org/10.1007/s00429-020-02122-6
REFERENCIAS:
Guy, J., et al. A. A mouse Mecp2-null mutation causes neurological symptoms that mimic rett syndrome. Nature Genetics, 27(3), 322–326. 2001. DOI: https://doi.org/10.1038/85899
Hagberg, B. Clinical manifestations and stages of rett syndrome. Mental Retardation and Developmental Disabilities Research Reviews, 8(2), 61–65. 2002. DOI: https://doi.org/10.1002/mrdd.10020
Otero-Garcia, Met al. Extending the socio-sexual brain: Arginine-vasopressin immunoreactive circuits in the telencephalon of mice. Brain Structure and Function. . 2014. DOI: https://doi.org/10.1007/s00429-013-0553-3
Rood, BD, et al. Site of origin of and sex differences in the vasopressin innervation of the mouse (Mus musculus) brain. Journal of Comparative Neurology, 521(10), 2321–2358. 2013. DOI: https://doi.org/10.1002/cne.23288
Tantra, M, et al. Mild expression differences of MECP2 influencing aggressive social behavior. EMBO Molecular Medicine. 2014. DOI: https://doi.org/10.1002/emmm.201303744
Zoghbi HY, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genetics. 1999. 23(2), 185–188. DOI: https://doi.org/10.1038/13810

El gen AtCDF3 promueve una mayor producción de az...
Un estudio con datos de los últimos 35 años, ind...
Un equipo de investigadores de la Universidad Juli...

En nuestro post hablamos sobre este interesante tipo de célula del si...

Regenera Activa Worldwide y Rigenera HBW, empresas biotecnológicas es...










