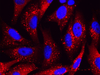
Las CNVs (Copy Number Variations) son un tipo de variantes estructurales de tamaño submicroscópico, como deleciones, duplicaciones, inserciones, inversiones y translocaciones, que implican un cambio en el número de copias respecto al genoma de referencia (Figura 1). Aunque son relativamente comunes en la estructura de nuestros genomas y contribuyen en gran medida a la variabilidad interindividual, también ha sido probada su implicación en un gran número de enfermedades como autismo, Parkinson, Alzheimer, enfermedades mitocondriales o cáncer, entre otras.
Las tecnologías Next-generation Sequencing (NGS) se han convertido en una herramienta clave para la identificación de CNVs, sustituyendo casi por completo a otras como los CGH arrays y MLPAs. Sin embargo, el análisis de los datos procedentes de la secuenciación de exomas presenta limitaciones que deben ser tenidas en cuenta, ya que pueden afectar a la precisión de los resultados:

Figura 1: Tipos de variaciones estructurales genómicas.
Aunque no existen unos estándares de referencia para el análisis de CNVs, siempre que sea posible se deben seguir una serie de recomendaciones alcanzar unos resultados óptimos:
La caracterización de CNVs sigue siendo complicada por lo que es necesario conocer y tener presentes sus limitaciones. Además, resulta clave llevar a cabo una correcta validación de aquellas variantes encontradas, especialmente en el entorno clínico. El desarrollo de bases de datos de acceso público, tanto en individuos controles como en pacientes, contribuirá en el futuro a dilucidar el verdadero significado de todas las CNVs analizadas.
BIBLIOGRAFÍA

El gen AtCDF3 promueve una mayor producción de az...
Un estudio con datos de los últimos 35 años, ind...
Un equipo de investigadores de la Universidad Juli...

En nuestro post hablamos sobre este interesante tipo de célula del si...

Horizon ha puesto en funcionamiento una nueva planta dedicada íntegra...