El escenario regulatorio de las Terapias Avanzadas es complejo. En él conviven autorizaciones de uso hospitalario con medicamentos plenamente aprobados por la EMA. Entender esta dualidad permite anticipar los requisitos operativos que diferencian ambas opciones.
Las terapias avanzadas (ATMP), que incluyen medicamentos de terapia génica, terapia celular y productos de ingeniería tisular, han cambiado el paradigma del tratamiento de enfermedades graves y poco frecuentes. Su complejidad tecnológica, sus requisitos de fabricación y la necesidad de garantizar un beneficio clínico robusto han dado lugar a un marco regulatorio singular en Europa y en España. En este contexto conviven dos figuras distintas: la autorización de uso hospitalario, limitada a un centro y a pacientes concretos, y la autorización de comercialización otorgada por la Agencia Europea del Medicamento (EMA) tras un procedimiento centralizado. Aunque ambas vías tienen como objetivo permitir el acceso controlado a terapias innovadoras, responden a necesidades clínicas y regulatorias muy diferentes.
Regulación aplicable en España y la Unión Europea
La Unión Europea regula los medicamentos de terapias avanzadas a través del Reglamento (CE) 1394/2007, que exige que cualquier ATMP destinado a comercialización sea evaluado mediante el procedimiento centralizado de la EMA. Paralelamente, la Directiva 2001/83/CE ofrece el marco general para medicamentos de uso humano. Dentro de este sistema armonizado, el Reglamento reconoce que los Estados miembros pueden autorizar el uso de terapias avanzadas no autorizadas en circunstancias excepcionales, siempre que su fabricación y utilización estén estrictamente controladas. Este mecanismo, conocido como Hospital Exemption, se articula en España a través de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS), que evalúa la petición caso por caso.
Qué es la autorización de uso hospitalario en terapias avanzadas
La autorización de uso hospitalario permite que una terapia avanzada no autorizada a escala europea pueda administrarse en un hospital concreto y únicamente para pacientes definidos. No constituye una autorización comercial, sino un permiso excepcional con finalidad estrictamente asistencial. Se utiliza cuando no existe alternativa terapéutica autorizada y la terapia propuesta podría aportar un beneficio clínico relevante. Su producción tiene lugar generalmente en unidades hospitalarias o centros autorizados, bajo condiciones de calidad GMP adaptadas a producciones no industriales o a pequeña escala. La AEMPS revisa el proceso, el control de calidad, la experiencia del centro, la justificación clínica y los mecanismos de farmacovigilancia aplicables en el ámbito local.
En España, hasta la fecha, solamente se han autorizado cinco terapias avanzadas, bajo este esquema, la primera de las cuales – productos celulares mesenquimales para la lesión medular traumática crónica – fue gestionada por Sermes CRO para la Fundación para Investigación del Hospital Universitario Puerta de Hierro. Todas ellas se dirigen a un hospital y a un perfil clínico específico, no se comercializan y no pueden transferirse libremente a otros centros.
Qué es la autorización de un medicamento de terapia avanzada por la EMA
Cuando una terapia avanzada aspira a comercializarse, debe someterse a un proceso regulatorio mucho más ambicioso. El procedimiento centralizado de la EMA exige un expediente completo con datos preclínicos, estudios clínicos de fase I, II y III, información exhaustiva de calidad y fabricación bajo condiciones GMP industriales, así como procesos de farmacovigilancia y un plan detallado de gestión de riesgos. La autorización otorgada tras esta evaluación es válida en toda la UE y permite distribuir el medicamento en cualquier Estado miembro, siempre conforme a las decisiones nacionales de financiación y acceso.
Se trata de medicamentos autorizados por la EMA y disponibles en Europa bajo indicaciones concretas, incluidos CAR-T (Yescarta, Kymriah, Tecartus, Breyanzi), terapias génicas como Luxturna o Zolgensma, productos para enfermedades hereditarias como Roctavian o Hemgenix, etc. Todos ellos sometidos a un proceso comparativamente más exigente que el de uso hospitalario y cuya fabricación depende de sistemas industriales altamente estandarizados.
Dos vías complementarias que responden a necesidades diferentes
La coexistencia de ambas figuras es necesaria porque cumplen funciones distintas. La autorización de uso permite tratar a un paciente o a un grupo muy reducido cuando la alternativa autorizada simplemente no existe. Ofrece una solución clínica inmediata, aunque limitada en su alcance. La autorización europea, en cambio, garantiza la disponibilidad homogénea del medicamento en toda la Unión Europea y se apoya en datos de eficacia mucho más robustos y reproducibles.
Para los equipos de investigación clínica, la diferencia no es solo conceptual sino operativa. Una terapia avanzada autorizada para uso hospitalario difícilmente puede integrarse en un ensayo clínico multicéntrico sin modificaciones regulatorias significativas. Un ATMP autorizado por la EMA, en cambio, sí puede formar parte del arsenal terapéutico estándar, ser comparador en estudios o integrarse en estrategias de tratamiento complejas. El desarrollo clínico también difiere: un producto que parte de una autorización de uso no puede escalar sin rediseñar su proceso de fabricación y sin completar un programa clínico completo si aspira a convertirse en medicamento.
Preguntas y respuestas: Terapias avanzadas, uso hospitalario y autorización europea
La autorización de uso hospitalario permite aplicar una terapia avanzada únicamente en un hospital concreto y para pacientes específicos. La autorización de comercialización, en cambio, permite el uso del medicamento en toda la UE tras superar un proceso regulatorio centralizado.
La AEMPS, tras evaluar la calidad del producto, el proceso de fabricación, la experiencia del centro y la justificación clínica.
No. Es asistencial y está limitada a un entorno hospitalario concreto.
Para obtener una autorización de comercialización por la EMA, un medicamento de terapia avanzada debe presentar un expediente completo de desarrollo, que incluya datos preclínicos y clínicos robustos, normalmente procedentes de estudios de fase I, II y III, demostrar de forma concluyente su eficacia y seguridad, disponer de un proceso de fabricación industrial validado conforme a GMP, y contar con un sistema integral de farmacovigilancia y un plan de gestión de riesgos aplicable a toda la Unión Europea.
Aunque parte de esta documentación también se exige en la autorización de uso hospitalario —especialmente en lo relativo a calidad, fabricación y justificación clínica—, en ese caso su alcance es más limitado, está orientado a un uso asistencial concreto y no sustituye al programa completo de desarrollo clínico requerido para la comercialización.
Debe presentar un expediente completo con estudios preclínicos y clínicos, demostrar eficacia y seguridad, cumplir GMP industriales y disponer de un plan de gestión de riesgos y farmacovigilancia.
Sí, pero solo si desarrolla un programa clínico completo y adapta su proceso de fabricación a los estándares industriales exigidos para los ATMP.
Porque la autorización de uso se concede a un hospital concreto, no a todos. Su disponibilidad depende de la capacidad del centro para fabricarla y de la autorización específica de la AEMPS.
Solo si se tramita un ensayo conforme al Reglamento (UE) 536/2014 a través de CTIS y la terapia cumple requisitos de fabricación, calidad y trazabilidad equivalentes a los exigidos en investigación clínica.
Analizar la vía regulatoria adecuada, coordinar los requisitos operativos, ayudar al promotor a planificar el desarrollo clínico y garantizar que la fabricación, documentación y estrategia regulatoria cumplen los estándares exigidos.
Artículo originalmente publicado en la revista PMFarma: Autorización de uso frente a autorización de medicamento en Terapias Avanzadas: diferencias clave desde la investigación clínica | Artículos PMFarma
Por Dolores Pérez Méndez, Quality Department Director en Sermes CRO

La mejor actitud que podemos adoptar es la de trat...

El equipo de investigadores observó cambios en el...

El gen AtCDF3 promueve una mayor producción de az...

En nuestro post hablamos sobre este interesante tipo de célula del si...
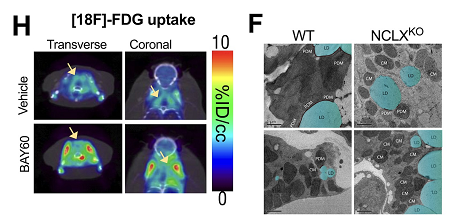
El estudio ha identificado un mecanismo fundamental que regula cómo e...