La Agencia Europea del Medicamento en su búsqueda por conseguir una armonización de la actuación de los departamentos de farmacovigilancia y facilitar el trabajo de las personas involucradas publicó las Good Vigilance Practices (GVP).
El módulo VI de las GVP hace referencia al modo en el que se deben gestionar los casos clínicos en un departamento de farmacovigilancia y la elaboración de los informes Individual Case Safety Report (ICSR). Fue publicado con el objetivo de establecer los requerimientos legales que conciernen a los Titulares de Autorización de Comercialización y a las Autoridades Nacionales Competentes dentro de la Unión Europea.
ICSR es el documento que informa sobre una o varias reacciones adversas (RA) experimentadas por un paciente en un momento específico.
Cuando nos enfrentamos a la gestión de un caso clínico, como punto de partida será necesario establecer si el caso es un efecto adverso (cualquier acontecimiento perjudicial para la salud que se presente en un paciente o sujeto de una investigación clínica al que se ha administrado un medicamento, aunque no tenga necesariamente una relación causal con dicho tratamiento) y una reacción adversa (respuesta nociva y no intencionada a un medicamento).
Para realizar este análisis de causalidad no existe un método estándar. El método de evaluación más comúnmente empleado en los departamentos de farmacovigilancia es la evaluación mediante algoritmos, siendo los más empleados el algoritmo de Karch y Lasagna modificado por Naranjo at col y el algoritmo de la OMS (WHO).
Se evaluarán también la gravedad del caso de acuerdo a los criterios establecidos en las GVPs y el listado Important Medical Events (IME) y la esperabilidad de acuerdo al documento de seguridad de referencia del medicamento.
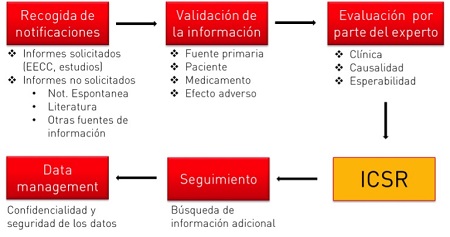
El proceso de gestión de un caso comienza con la recogida de notificaciones por parte del experto en farmacovigilancia, que han de ser perfectamente documentadas y clasificadas en el sistema de farmacovigilancia. Las notificaciones provienen principalmente de dos fuentes, solicitadas (provenientes de Ensayos Clínicos, programas de apoyo a pacientes…) o no solicitadas (notificaciones publicadas en revistas científicas, medios de comunicación, provenientes de profesionales sanitarios o pacientes/consumidores…).
La información mínima para validar un caso y poder por tanto gestionarlo adecuadamente, es la siguiente:
Sin uno de estos cuatro datos, la notificación no puede considerarse válida.
Toda la información de seguridad recogida en un departamento de farmacovigilancia es extremadamente sensible, por lo que ha de tratarse con máxima confidencialidad y seguridad. Además, se debe establecer un sistema de calidad en todas las fases del proceso que asegure que se cumplen con los estándares exigidos.
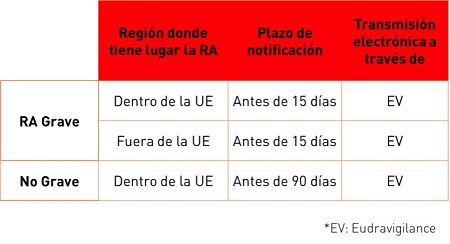
El envío expeditivo de los casos de forma electrónica es obligatorio en todos los países de la Unión Europea. Este envío se deberá realizar mediante la base de datos Eudravigilance; los plazos para el envío de los casos son los siguientes:

La mejor actitud que podemos adoptar es la de trat...

El equipo de investigadores observó cambios en el...

El gen AtCDF3 promueve una mayor producción de az...

En nuestro post hablamos sobre este interesante tipo de célula del si...

Un nuevo estudio relaciona las temperaturas elevadas en la primera inf...










