Un artículo publicado por investigadores del Instituto de Química Teórica y Computacional de la UB (IQTCUB) ha sido seleccionado por la revista The Journal of Chemical Physics (JCP) como artículo Editors’ Choice 2017. La colección Editors’ Choice 2017 (selección de los editores 2017) de JCP tiene 73 artículos, que los editores han escogido por ser los «más innovadores y con mayor impacto de 2017».
El artículo, que se publicó en agosto de 2017, es el resultado de una colaboración de los investigadores del IQTCUB Josep M. Bofill, Jordi Ribas Ariño y Sergio Pablo García con el Instituto de Matemáticas de la Universidad de Leipzig. El objeto del estudio era la creación de un algoritmo para determinar tanto la formación como la ruptura por estrés de enlaces moleculares para la síntesis y catálisis químicas.
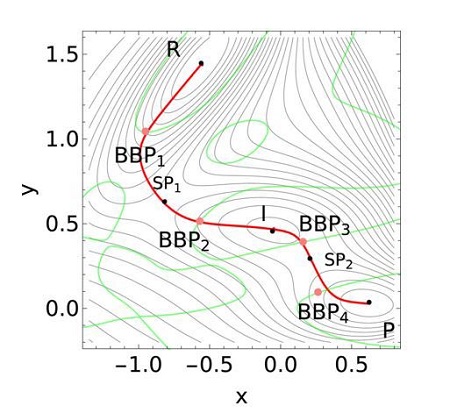
Los últimos avances en microscopia de fuerzas atómicas han permitido a los investigadores aplicar fuerzas mecánicas a moléculas individuales para provocar reacciones químicas. Este equipo de investigadores ha desarrollado el primer algoritmo de este tipo, que determina la fuerza mínima que se necesita para alcanzar el mejor punto de ruptura (bond-breaking point BBP) a nivel molecular para provocar una reacción química mecánicamente.
«El algoritmo se puede aplicar a cualquier molécula, incluidas moléculas biológicas como las proteínas, y también moléculas inorgánicas y orgánicas», explica Josep M. Bofill, jefe del grupo del IQTCUB. Esta investigación tiene implicaciones para muchas aplicaciones, como las máquinas moleculares, los polímeros mecánicamente resistentes y autorregenerables, los materiales resistentes a las tensiones y el diseño de catalizadores. El algoritmo también puede utilizarse para explorar el modo en que los campos eléctricos externos pueden catalizar y controlar las reacciones químicas.
La flexión y la torsión de los enlaces de una molécula tienen una rigidez variable. Por tanto, determinar el apoyo que aguanta la fuerza de una molécula, para predecir, por ejemplo, el punto de ruptura del enlace en una molécula sobretensionada, implica que se deben analizar las distintas direcciones de la fuerza externa.
El algoritmo está basado en las trayectorias de Newton, que proceden del método matemático para calcular los ceros de una función. En el caso de los puntos de ruptura de los enlaces, las trayectorias de Newton se situarían cerca del camino de la reacción química.
Artículo de referencia:
J. M. Bofill, J. Ribas-Ariño, S. Pablo García, W. Quapp. «An algorithm to locate optimal bond breaking points on a potential energy surface for applications in mechanochemistry and catalysis». The Journal of Chemical Physics 147, 152710 (2017). Doi: 10.1063/1.4994925

La mejor actitud que podemos adoptar es la de trat...

El equipo de investigadores observó cambios en el...

El gen AtCDF3 promueve una mayor producción de az...

En nuestro post hablamos sobre este interesante tipo de célula del si...

Un artículo publicado en Alzheimer’s & Dementia: The Journal of the...










