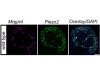
Hacía años que se sabía que mutaciones en las dos copias del gen HERC2 se asocian a un trastorno del neurodesarrollo que provoca retraso global del desarrollo, discapacidad intelectual, rasgos del espectro autista y alteraciones del movimiento. Unas características muy similares al síndrome de Angelman, más conocida pero también minoritaria. Sin embargo, no estaba clara cuál es la función exacta del gen afectado (HERC2) y con qué moléculas interactúa. Sin conocer la biología detrás del síndrome, era difícil entender cómo funciona y plantear estrategias terapéuticas para tratarlo.
Ahora, un estudio científico liderado por investigadores del Instituto de Investigación Biomédica de Bellvitge (IDIBELL) y de la Universidad de Barcelona, en el Campus Salud Bellvitge, ha profundizado en el papel del gen HERC2 en trastornos del neurodesarrollo de este tipo y ha identificado su papel clave en la degradación selectiva de proteínas —el «control de calidad» de las proteínas dentro de la célula—, que se ejecuta a través del sistema del proteasoma. El trabajo, publicado en la revista Cell Death Discovery, ha sido dirigido por José Luis Rosa y cuenta con Joan Sala-Gaston y Laura Costa-Sastre como primeros autores, y significa los primeros pasos para descifrar la biología de trastornos causados por este gen.
Una familia de genes vinculada a diferentes trastornos del neurodesarrollo
Las variantes patogénicas en los genes HERC1 y HERC2, que codifican enzimas de tipo E3 ligasa de ubiquitina, se han asociado a diferentes enfermedades del neurodesarrollo. En el caso de HERC1, las alteraciones genéticas en ambas copias del gen causan un síndrome minoritario caracterizado por macrocefalia, rasgos faciales dismórficos y retraso psicomotor. En el caso de HERC2, las variantes en ambas copias del gen que reducen su función se asocian a un trastorno del neurodesarrollo con características similares al síndrome de Angelman, que incluyen retraso global del desarrollo, discapacidad intelectual, rasgos del espectro autista y alteraciones del movimiento. A pesar de esta relación con diversas patologías, sin embargo, hasta ahora se conocía poco sobre qué proteínas regula HERC2, y por lo tanto, cómo estas alteraciones contribuyen a la enfermedad.
Para resolver esta cuestión, los investigadores han utilizado técnicas avanzadas de proteómica cuantitativa que permiten identificar las proteínas marcadas por ubiquitina —una señal que en muchos casos dirige a las proteínas hacia su degradación—. Mediante esta aproximación, se han identificado numerosas proteínas de complejos proteicos regulados por HERC2 importantes para el funcionamiento celular, como el proteasoma y las maquinarias de síntesis de tRNA, de inicio de la traducción, del transporte vesicular, de formación de centrosomas y del citoesqueleto, entre otros.
Error identificado: el control de calidad proteico falla
Entre todos estos sistemas, el correcto funcionamiento del proteasoma destaca como el principal objetivo funcional de HERC2. Los resultados muestran que la proteína HERC2 reconoce subunidades proteicas que aún no se han ensamblado correctamente y las marca para su eliminación, un proceso que depende de la interacción con proteínas tipo chaperonas. «Básicamente, la función de HERC2 es detectar las proteínas defectuosas y marcarlas con una etiqueta (ubiquitina) que indica que no están bien montadas, y por lo tanto, hay que degradarlas», explica el Dr. Jose Luís Rosa, investigador principal del grupo de Señalización celular y biología del hueso del IDIBELL y la UB. Este mecanismo es esencial para garantizar un ensamblaje adecuado del proteasoma y, sobre todo, mantener el equilibrio de proteínas dentro de la célula. «Por eso, cuando HERC2 no funciona correctamente, este equilibrio se ve alterado, acumulando proteínas que habría que degradar, y se afecta la actividad del proteasoma«, añade.
Estas conclusiones se han visto reforzadas por experimentos realizados en células derivadas de pacientes que llevan una variante patogénica frecuente del gen HERC2. «Las muestras de los pacientes mostraban exactamente los mismos problemas en el sistema de degradación de proteínas, y una actividad anómala del proteasoma», remarca Joan Sala-Gaston, investigador postdoctoral y primer autor del estudio.
En conjunto, el estudio establece un vínculo directo entre las alteraciones del gen HERC2 y el funcionamiento del sistema de degradación de proteínas, aportando nuevos conocimientos sobre los mecanismos moleculares que pueden originar los trastornos del neurodesarrollo que se observan en los pacientes. En este sentido, el trabajo se enmarca en la apuesta del IDIBELL por reforzar la investigación en patologías poco frecuentes a través del programa REMMA Bellvitge (Investigación en Enfermedades Minoritarias del Adulto de Bellvitge). Aunque todavía queda camino por recorrer, «Entender las bases biológicas de este síndrome minoritario es el paso necesario para poder diseñar, en un futuro, estrategias terapéuticas capaces de restaurar el equilibrio de las proteínas y mejorar la vida de los pacientes«, concluye Laura Costa, investigadora predoctoral y primera autora del estudio.

La mejor actitud que podemos adoptar es la de trat...

El equipo de investigadores observó cambios en el...

El gen AtCDF3 promueve una mayor producción de az...

En nuestro post hablamos sobre este interesante tipo de célula del si...
Un estudio liderado por la URV, el IRB-CatSud y el CIBEROBN identifica...