Joan J. Guinovart’s lab at the Institute for Research in Biomedicine (IRB Barcelona) forms part of a recently launched international consortium that seeks to find a treatment for Lafora disease—the most severe type of epilepsy in humans.
Headed by the University of Kentucky (US), the project has received $8.5 M (7.7 M€) of funding from the National Institutes of Health (NIH) for the next five years. The project took shape with the help of Chelsea’s Hope, a charity organisation formed by families in the US affected by Lafora disease and that, in 2014, brought together leading experts working in different aspects of this disease.
Lafora disease is a hereditary neurodegenerative condition in which the first epileptic seizure occurs during adolescence, between the age of 10 and 17 and for which no treatment is available. The disease involves the progressive degeneration of the nervous system, with patients deteriorating into a vegetative state and dying some ten years after its onset.
The research team comprises labs headed by Matthew Gentry, at the University of Kentucky —leader of the consortium who will be supported by Pascual Sanz at the Instituto de Biomedicina de Valencia of the CSIC—, by Berge Minassian at the Hospital for Sick Kids in Toronto, Canada, by Peter Roach at the University of Indiana, in the US, by José Serratosa at the Hospital Universitario Fundación Jiménez Díaz de Madrid, and by Joan Guinovart at IRB Barcelona.
“We have all been studying Lafora disease for over ten years and we have come together to work towards a breakthrough. It is great that the initiative arose from families,” highlights Joan Guinovart, who is also a professor at the University of Barcelona. “Only by joining forces can we find solutions to rare diseases like the one we are dealing with,” he says.
What do we know about Lafora disease?
“It is the first type of epilepsy for which the molecular bases are known,” explains Guinovart. It is transmitted when both parents pass on a mutation in one of the two genes involved with the disease, namely laforin (named after Dr. Lafora, a Spanish neurologist who discovered the disease) and malin (from the French term le grand mal used to refer to epilepsies). The disease is characterised by the accumulation of abnormal deposits, called Lafora bodies, in neurons.
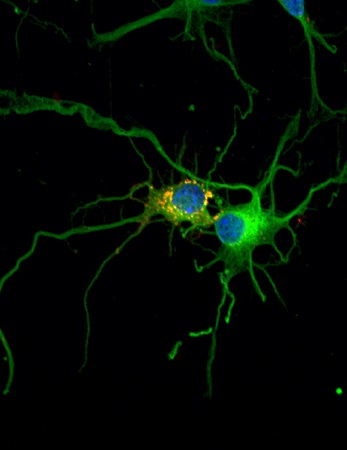
” In an article published in Nature Neuroscience in 2007, Guinovart and the members of his team, experts in glycogen metabolism, demonstrated that mutations in either of these two genes lead to the accumulation of glycogen—chains of sugar—in neurons, thus causing toxicity and triggering neuronal death. Likewise, using mice, they showed that the inhibition of glycogen synthesis in neurons prevents the development of Lafora disease, thereby identifying a possible treatment for this condition.
“We now have to study whether blocking glycogen synthesis after the onset of the disease can halt its progression or even reverse the condition. We are also looking into ways to prevent the disease,” he says. Each lab will focus on different aspects of the disease, covering from basic to translational research, with the aim to make breakthroughs that can be used to design a clinical assay involving patients.
Image: Confocal microscopy image. The accumulation of glycogen (yellow and red) in neurons leads to their deterioration and finally death (IRB Barcelona)

La mejor actitud que podemos adoptar es la de trat...

The research team observed changes in head circumf...

AtCDF3 gene induced greater production of sugars a...

En nuestro post hablamos sobre este interesante tipo de célula del...

Telum Therapeutics, a biotechnology company leveraging proprietary met...