Un equipo de investigación, formado por personal del Departament de Biologia Celul·lar i Biologia Funcional de la Universitat de València (UV), la Unitat Predepartamental de Medicina de la Universitat Jaume I de Castelló (UJI) y la Queen Mary University of London (QMUL), ha mostrado que la mutación del gen Mecp2 altera el funcionamiento del eje hipotálamo-hipofisario-gonadal, responsable del control de los niveles de hormonas sexuales, y retrasa el desarrollo puberal en ratones modelo del síndrome de Rett.
El síndrome de Rett es una enfermedad rara del neurodesarrollo, casi siempre letal en el sexo masculino, que se manifiesta casi exclusivamente en niñas y mujeres. Este síndrome provoca síntomas muy graves, como epilepsia, discapacidad intelectual y motora o dificultades respiratorias. Además de estos síntomas, la enfermedad afecta al sistema neuroendocrino, pero este aspecto ha recibido menos atención.
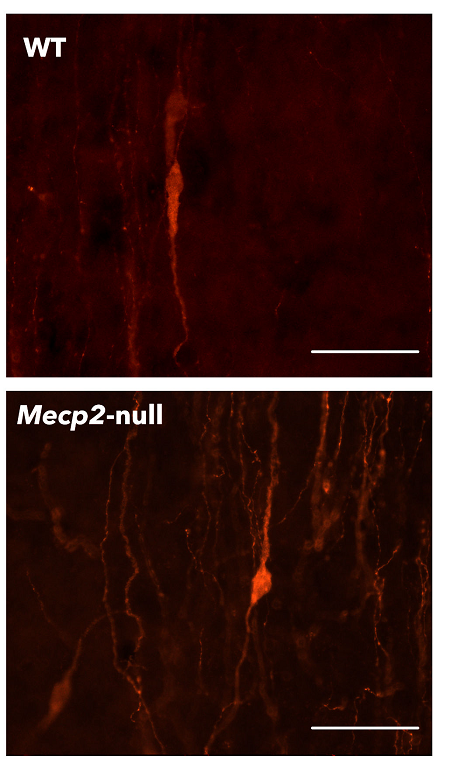
Ahora, un equipo internacional dirigido por Carmen Agustín Pavón, profesora de la Facultad de Ciencias Biológicas de la UV, ha descubierto que en ratones mutantes para el gen Mecp2, causa principal del síndrome de Rett, hay un exceso de neuronas GnRH localizadas en el hipotálamo, unas células cuya señalización es responsable del inicio de la pubertad y la regulación de las hormonas esteroides sexuales. No obstante, el desarrollo puberal de los ratones mutantes se encuentra retrasado con respecto a sus congéneres de la misma edad, asociado a su bajo peso, y los niveles de hormonas sexuales en ratones mutantes son más bajos que en individuos sanos, sugiriendo un mal funcionamiento del denominado eje hipotálamo-hipofisario-gonadal.
«Sabíamos que en algunas pacientes con síndrome de Rett la pubertad sigue un curso atípico, que puede cursar con un inicio precoz pero una primera menstruación retrasada», comenta Ana Martín-Sánchez, de la Universitat Jaume I de Castelló y primera firmante del estudio. «El hecho de que el sistema de control de las hormonas sexuales se encuentre alterado no solo impacta sobre la pubertad y los ciclos menstruales, sino que estas hormonas son imprescindibles para la organización y el mantenimiento de los circuitos neurales que controlan el comportamiento social, también afectados en estos ratones, la salud cognitiva y la del sistema musculoesquelético, entre otras funciones», continúa la investigadora de la UJI.
«Conocer en detalle los mecanismos por los que la falta de MeCP2 da lugar a alteraciones neuroendocrinas nos podría abrir la puerta a ensayar terapias de sustitución hormonal, que ya se utilizan de rutina para otras condiciones, para pacientes con Rett, lo que podría redundar en una mejora de su calidad de vida», concluye Carmen Agustín Pavón.
Además del equipo de la UV y la UJI, el trabajo se ha realizado en colaboración con Sasha R. Howard, endocrinóloga pediátrica del Hospital Barts Health NHS Trust y la QMUL. La investigación ha sido financiada por una subvención para grupos de investigación consolidados de la Conselleria de Educación, Cultura y Universidades de la Generalitat Valenciana (CIAICO/2023/027) y por el Fondo para la Investigación para el Síndrome de Rett (FinRett) impulsado por las Asociaciones Española y Catalana de síndrome de Rett.
Artículo: Martín-Sánchez, A.; Jiménez-Díaz, D.; Esteve-Pérez, R.; Vasile-Tudorache, A.; Read, JE.; Howard, SR.; Agustín-Pavón, C. “Pubertal development and hypothalamic-pituitary-gonadal axis are altered in male mice lacking Mecp2”. J Neuroendocrinol. 2026 Jul;38(7):e70221. https://doi.org/10.1111/jne.70221
Imagen: Fotomicrografía de fluorescencia de neuronas GnRH de un ratón sano (WT) y de otro enfermo (Mecp2-null).

La mejor actitud que podemos adoptar es la de trat...

El equipo de investigadores observó cambios en el...

El gen AtCDF3 promueve una mayor producción de az...

En nuestro post hablamos sobre este interesante tipo de célula del si...
Un estudio del CNIC, CSIC e IIS-FJD ha logrado visualizar la acumulaci...