
Del total de ADN que compone nuestro genoma, entre un 95-98 % corresponde a pseudogenes, intrones, secuencias UTR, y otros ADNs no codificantes. En humanos, se estima que existen más 19.000 pseudogenes con secuencias semejantes a las de genes conocidos, pero sin regiones esenciales para la codificación de proteínas. Aunque se ha asumido que son el resultado de la pérdida de función de algunos genes en el curso de la evolución, hoy en día se sabe que algunos desempeñan funciones importantes para nuestro organismo. Los pseudogenes se clasifican en dos categorías principales:
– Unitarios: son el resultado de la acumulación de una serie de mutaciones de diferentes tipos en un gen codificante cuya consecuencia es la pérdida de funcionalidad total o parcial de éste. En este caso, por lo tanto, no existe un gen homólogo funcional.
– Duplicados: son el resultado de la duplicación de un gen funcional. El proceso de duplicación puede no ser completo, de manera que la copia carezca de algunas regiones y por lo tanto, no sea funcional. También puede ocurrir que el gen original o el duplicado acumulen mutaciones que den lugar a la pérdida de funcionalidad de uno de ellos.
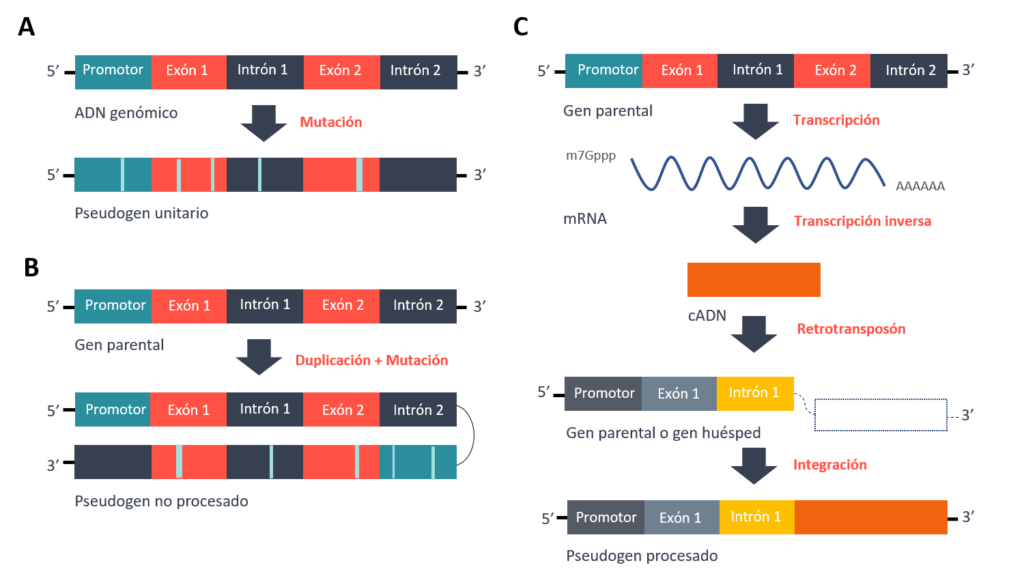
Tipos de pseudogenes: (A) Pseudogenes unitarios, resultado de la acumulación de mutaciones. (B) Pseudogenes duplicados, resultado de la duplicación de un gen funcional. (C) Pseudogenes procesados, resultado de un proceso de transcripción reversa.
Imagen adaptada del artículo: Theranostics 2020; 10(4):1479-1499. doi:10.7150/thno.40659.
El alto grado de homología compartido entre algunos pseudogenes con regiones funcionales del genoma supone un importante problema cuando se llevan a cabo análisis genómicos con enfoques Next-generation Sequencing (NGS), especialmente en el entorno clínico. Las secuencias pseudogénicas se caracterizan por tener unas tasas de mutación mucho más altas, por lo que su mapeo en el correspondiente gen funcional puede ocasionar falsos positivos, y el caso contrario, en el que lecturas provenientes del gen funcional con mutaciones mapeen en un pseudogen, puede dar lugar a falsos negativos. Por lo tanto, la secuenciación de pseudogenes puede afectar a la interpretación de las mutaciones identificadas como causantes de una patología. Aunque las herramientas bioinformáticas reducen notablemente este inconveniente, es aconsejable realizar un abordaje específico cuando se conoce la presencia de pseudogenes.
A continuación, se ofrecen algunas recomendaciones para contrarrestar las limitaciones tecnológicas y aumentar la fiabilidad de los resultados:
En resumen, la aplicación de la tecnología NGS en el diagnóstico clínico requiere de una particular atención para distinguir los pseudogenes de las regiones funcionales del genoma. Sin embargo, no hay que perder de vista la posible contribución de las secuencias pseudogénicas a la biología de nuestro organismo. En el futuro, los avances en tecnologías NGS y análisis bioinformático ayudarán a esclarecer sus funciones y a entender su papel en el desarrollo de algunas enfermedades.
BIBLIOGRAFÍA

La mejor actitud que podemos adoptar es la de trat...

The research team observed changes in head circumf...

AtCDF3 gene induced greater production of sugars a...

En nuestro post hablamos sobre este interesante tipo de célula del...

New study finds an association between higher temperatures early in li...










