El síndrome de Rett (RTT, MIM 312750) es una enfermedad neurológica muy severa, con un desorden progresivo del neurodesarrollo, constituye la segunda causa de retraso mental en mujeres y tiene una incidencia de 1:10000-15000 nacimientos. Fue descrita por primera vez en por Andreas Rett en 1966 y se manifiesta después de un período de desarrollo de aparentemente normalidad entre los 6-18 meses de edad, con una posterior desaceleración del crecimiento, deterioro psicomotor severo y retraso mental. Por tanto, los pacientes RTT tienen pérdidas de habilidades cognitivas, sociales y motoras, gradualmente pierden el habla, desarrollan microcefalias, convulsiones, autismo, hiperventilación y, como consecuencia, tienen una disminución de la esperanza de vida. La terapia actual es solo paliativa de algunos de los síntomas puesto que no existe una cura eficaz y es por ello que representa una enfermedad devastadora para las pacientes y para las familias. Por lo tanto, se hace muy necesario profundizar en los mecanismos moleculares asociados al desarrollo del síndrome de Rett, con el objetivo de encontrar una terapia curativa.
En este sentido, uno de los avances más importantes en la investigación del síndrome de Rett fue el descubrimiento de mutaciones en el gen que codifica para la proteína MeCP2 (methyl-CpG-binding protein 2) y su asociación al desarrollo de la enfermedad.
MECP2 es una proteína nuclear que se expresa abundantemente en el sistema nervioso y entre sus funciones principales se ha descrito la capacidad de unirse a regiones metiladas del ADN consiguiendo de esta manera controlar la expresión génica.
Modelo murino del síndrome de Rett
En el presente trabajo se utilizó el modelo murino descrito por Guy y col., 2001 (B6.129P2(c)-Mecp2tm1+1Bird) en el laboratorio del Dr Adrian Bird. Estos ratones tienen un comportamiento normal hasta alcanzar las 4 semanas de edad. A partir de ahí hay una degeneración motora y cognitiva que va acompañada de la aparición de síntomas similares a los del síndrome de Rett en humanos.
El estudio se ha encaminado a profundizar en las vías de señalización molecular de Gsk3b (Glycogen sinthase kinase 3b) dado que esta proteína tiene un papel crucial en el desarrollo neural.
Han sido descritos dos residuos aminoacídicos que regulan su actividad kinasa, dígase la capacidad de fosforilar a otras proteínas y activar diversas señalizaciones moleculares. Estos residuos son Ser9 y Tyr216. La fosforilación del primero está relacionada con la inhibición de la actividad enzimática mientras que el segundo lo está con la activación.
El tratamiento con SB216763, un inhibidor de Gsk3b, disminuye la actividad enzimática in vitro y aumenta la supervivencia en un modelo murino del síndrome de Rett
El ARN de Gsk3b se halla ampliamente distribuido en el cerebro de ratones a la edad de 8 semanas. Sin embargo, en este estudio los autores demostraron que la actividad enzimática está aumentada en el cerebelo de los ratones deficientes para la proteína Mecp2 y también en muestras cerebrales de pacientes con síndrome de Rett. Estas observaciones fueron confirmadas en cultivos primarios de neuronas, mediante técnicas de inmunomarcajes para los aminoácidos Ser9 y Tyr216. Es de destacar que el tratamiento de los cultivos con SB216763 disminuyó la actividad enzimática de Gsk3b a los niveles de los ratones sin mutación en Mecp2.
De manera similar el tratamiento con este mismo inhibidor disminuyó lo síntomas típicos del síndrome de Rett en el modelo murino utilizado. Hubo una significante mejora en la movilidad y en la respiración, lo que conllevó a un aumento del bienestar animal y de la supervivencia.
Neuroinflamación en el síndrome de Rett
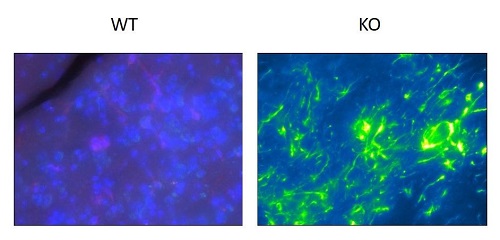
Es válido señalar que un aumento de la actividad enzimática de Gsk3b está muy relacionado con los procesos de inflamación y de estrés oxidativo, además de que hay numerosos estudios que apoyan esta hipótesis debido en parte al aumento de Nfkb1, una molécula clave en la regulación de la inflamación. Las evidencias encontradas en este estudio refuerzan estas afirmaciones ya que se encontraron niveles elevados de Nfkb1 en los cultivos de neuronas primarias provenientes de ratones deficientes para Mecp2, así como en el cerebelo del mismo modelo murino. También se observó que los niveles de Nfkb1 y de citoquinas proinflamatorias disminuyeron después del tratamiento con el inhibidor de Gsk3b. Otro parámetro a evaluar fue la sobreexpresión de Cd11b, (marcador de microglía en los ratones Rett). Se confirmó una elevada infiltración de estas células proinflamatorias en el cerebelo, que disminuyó sustancialmente tras el tratamiento con el inhibidor.
El tratamiento con el inhibidor recupera parcialmente la red neuronal, la espinogénesis y la actividad neuronal in vitro e in vivo
Se ha descrito que en muestras cerebrales de individuos post-mortem, la red dendrítica y la espinogénesis están reducidas. Sin embargo, en este trabajo el efecto del tratamiento con el inhibidor de Gsk3b conllevó una recuperación del árbol dendrítico y una mejora en la formación de las espinas.
Los resultados de este trabajo, liderado por el Dr. Manel Esteller y la Dra. Sonia Guil, sugieren que la activación de la proteína Gsk3b juega un papel importante en el síndrome de Rett y refuerzan el potencial terapéutico de la inhibición de esta vía de señalización para conseguir una disminución de la sintomatología y la activación de redes neuronales en un modelo preclínico del síndrome de Rett.
Se demuestra además la importancia de la inflamación en esta patología y la reducción de los efectos dañinos como consecuencia del efecto terapéutico del inhibidor, así como también la importancia de regular la vía de señalización donde interviene Gsk3b para regular los eventos neuronales excitatorios.
Es válido señalar que varios inhibidores de Gsk3b similares a SB216763 han sido estudiados como agentes terapéuticos en enfermedades como el Parkinson y el Alzheimer. Los resultados presentados en este estudio potencian la aplicabilidad de estos compuestos en otra patología neuronal como es el síndrome de Rett, por lo que se hace evidente profundizar en el análisis de esta vía de señalización molecular e investigar los mecanismos implicados en aras de una solución clínica para los pacientes afectados.
Referencia: Jorge-Torres et al. Inhibition of Gsk3b Reduces Nfkb1 Signaling and Rescues Synaptic Activity to Improve the Rett Syndrome Phenotype in Mecp2-Knockout Mice. Cell Reports, 2018, 23, 1665–1677. https://doi.org/10.1016/j.celrep.2018.04.010.
Bibliografía:
Guy, J. et al. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome Nat. Genet, 2001, 27, 322–326.doi:10.1038/85899.
Rett, A. On a unusual brain atrophy syndrome in hyperammonemia in childhood]., Wien. Med. Wochenschr. , 116, 723–726.
Fuente: Un nuevo fármaco demuestra eficacia preclínica en el síndrome de Rett. http://www.idibell.cat/modul/noticias/es/1076/un-nuevo-farmaco-demuestra-eficacia-preclinica-en-el-sindrome-de-rett
Imagen: Infiltración de células de la glía, marcadores de neuroinflamación en el modelo murino de síndrome de Rett en KO (deficientes para la proteína Mecp2) y WT (ratones salvajes para la proteína Mecp2). Imagen Jorge-Torres OC, Casal Moreno C. microscopio ZEISS. PEBC, Idibell.
Olga de la Caridad Jorge Torres1 2, Sonia Guil Domenech1,2
1 Programa de Epigenética y Biología del Cáncer (PEBC),
2 Instituto de Investigaciones Biomédicas de Bellvitge (IDIBELL), L’Hospitalet, Barcelona. España

La mejor actitud que podemos adoptar es la de trat...

The research team observed changes in head circumf...

AtCDF3 gene induced greater production of sugars a...

En nuestro post hablamos sobre este interesante tipo de célula del...

New study finds an association between higher temperatures early in li...










