A research team comprising researchers from the Department of Cell Biology and Functional Biology at the Universitat de València (UV), the Pre-Departmental Unit of Medicine at the Universitat Jaume I of Castelló (UJI), and Queen Mary University of London (QMUL) has shown that mutation of the Mecp2 gene disrupts the function of the hypothalamic-pituitary-gonadal axis, which regulates sex hormone levels, and delays pubertal development in a mouse model of Rett syndrome.
Rett syndrome is a rare neurodevelopmental disorder that is almost always lethal in males and affects almost exclusively girls and women. It causes severe symptoms, including epilepsy, intellectual and motor disabilities, and respiratory difficulties. In addition to these manifestations, the disorder also affects the neuroendocrine system, although this aspect has received comparatively little attention.
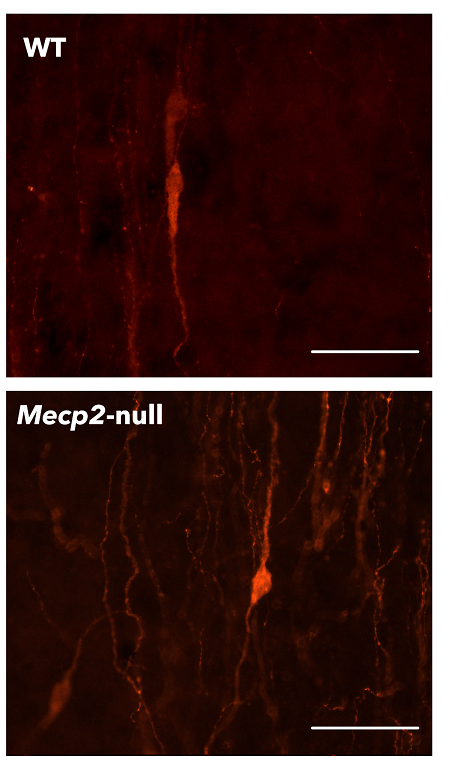
An international team led by Carmen Agustín Pavón, associate professor at the Faculty of Biological Sciences of the Universitat de València, has now discovered that mice carrying a Mecp2 mutation, the main cause of Rett syndrome, have an increased number of gonadotropin-releasing hormone (GnRH) neurons in the hypothalamus. These neurons are responsible for initiating puberty and regulating sex steroid hormone production. Despite this increase, pubertal development in the mutant mice is delayed compared with healthy mice of the same age, a delay associated with their lower body weight. In addition, mutant mice show lower sex hormone levels than healthy animals, suggesting impaired function of the hypothalamic-pituitary-gonadal axis.
"We already knew that puberty follows an atypical course in some patients with Rett syndrome, with an early onset but delayed menarche", explains Ana Martín-Sánchez, a researcher at the Universitat Jaume I and first author of the study. "Disruption of the system that regulates sex hormones not only affects puberty and menstrual cycles, but these hormones are also essential for the development and maintenance of the neural circuits involved in social behaviour, which are likewise altered in these mice, as well as for cognitive function and musculoskeletal health, among many other physiological processes", she adds.
"Understanding in detail how the lack of MeCP2 leads to neuroendocrine alterations could open the door to testing hormone replacement therapies, which are already routinely used for other conditions, in patients with Rett syndrome. This could ultimately contribute to improving their quality of life", concludes Carmen Agustín Pavón.
In addition to researchers from the Universitat de València and Universitat Jaume I, the study was carried out in collaboration with Sasha R. Howard, a paediatric endocrinologist at Barts Health NHS Trust and Queen Mary University of London. The research was funded by a grant for consolidated research groups awarded by the Regional Ministry of Education, Culture and Universities of the Valencian Government (CIAICO/2023/027) and by the Rett Syndrome Research Fund (FinRett), promoted by the Spanish and Catalan Rett Syndrome Associations.
Article: Martín-Sánchez, A.; Jiménez-Díaz, D.; Esteve-Pérez, R.; Vasile-Tudorache, A.; Read, J. E.; Howard, S. R.; Agustín-Pavón, C. “Pubertal development and hypothalamic-pituitary-gonadal axis are altered in male mice lacking Mecp2”. J Neuroendocrinol. 2026 Jul;38(7):e70221. https://doi.org/10.1111/jne.70221
Image: Fluorescence photomicrography of GnRH neurons of a healthy mouse (WT) and of another patient (Mecp2-null).

La mejor actitud que podemos adoptar es la de trat...

The research team observed changes in head circumf...

AtCDF3 gene induced greater production of sugars a...

En nuestro post hablamos sobre este interesante tipo de célula del...