Hace unos años, Javier Benítez, director del grupo de Genética Humana del Centro Nacional de Investigaciones Oncológicas (CNIO), fue contactado por Pablo García Pavía, del Servicio de Cardiología del Hospital Universitario Puerta de Hierro. Este cardiólogo estaba tratando a dos hermanos con una rara forma de cáncer, un angiosarcoma cardiaco (CAS).
"En aquella época probamos algunas ideas, pero sin éxito", explica Benítez. Ha habido que esperar a las modernas técnicas de análisis del genoma para descubrir el problema genético de los hermanos. El hallazgo abre una vía para identificar familias con CAS portadoras de una alteración en el gen responsable, cuyos miembros podrían beneficiarse de un diagnóstico precoz y de un tratamiento adecuado.
Los investigadores del grupo de Benítez retomaron el caso de los hermanos con CAS recientemente. Tras secuenciar su exoma —la parte del genoma que se traduce a proteínas y por tanto la que más influye en el estado del organismo—, hallaron la causa de su enfermedad en una mutación en un gen llamado POT1.
La identificación de este gen les conducía directamente a otro grupo del CNIO, el de Telómeros y Telomerasa, liderado por María Blasco. POT1 es una de las proteínas componentes del escudo protector de los telómeros —las estructuras que protegen los extremos de los cromosomas—, y recientemente ha sido identificado como responsable de otras formas de cáncer hereditario: el melanoma y el glioma familiar.
El grupo de Blasco no es solo uno de los grupos líderes en el estudio de los telómeros, sino que ya había participado en la primera descripción de la mutación de este gen en cáncer humano (leucemia linfocítica crónica) junto a los grupos de Carlos López-Otín y Elías Campo.
El angiosarcoma cardiaco es una enfermedad poco frecuente pero maligna. En los casos de CAS hereditario la supervivencia media es de solo cuatro meses, porque la enfermedad se diagnostica en fase avanzada. No se había identificado —hasta ahora— ningún gen implicado.
Los investigadores del CNIO observaron además que el CAS hereditario aparece en familias con una muy alta incidencia de otros tumores. Eso es parecido a lo que ocurre en los afectados por el síndrome Li-Fraumeni, causado por una mutación en el gen supresor de tumores, apodado guardián del genoma, P53. Pero las familias con CAS no tenían mutado P53, sino POT1.
Aplicación clínica inmediata
El hallazgo de la nueva mutación cobraba así una importancia aún mayor para la clínica, dado que señalaba a portadores en riesgo de padecer angiosarcoma cardiaco y también, posiblemente, otros tumores.
Como explica Benítez, "antes simplemente no había nada que ayudara a identificar a estas personas en riesgo, porque no había ningún marcador para CAS familiar ni para familias con síndrome similar a Li-Fraumeni sin mutaciones en P53. Este estudio descifra uno de los genes que explican la alta incidencia de cáncer en algunas de ellas".
"La traslación de estos resultados a la clínica es inmediata", afirma Blasco. "De hecho ya estamos ayudando a familias que portan la mutación".
En la consulta de cáncer familiar del CNIO que dirige Miguel Urioste, en el Hospital de Fuenlabrada, se analizan ya cerca de una decena de familias. También se ha creado una Unidad de Seguimiento de portadores asintomáticos en el Hospital Universitario Puerta de Hierro. El objetivo es ofrecer a estas personas asesoramiento genético y seguimiento, para detección precoz y tratamiento.
Los investigadores esperan que este hallazgo permita encontrar a más portadores que podrían no conocer su riesgo. Para ello han contactado con grupos y centros nacionales e internacionales implicados en la investigación de cáncer familiar en general y cardiogenéticos en particular, para notificarles el hallazgo y ampliar el estudio con más casos y familias.
La información debe llegar también a oncólogos, cardiólogos, genetistas y patólogos para que alerten y envíen muestras de los casos que encuentren entre sus pacientes.
Secuencia esencial para el cromosoma
Esta investigación, publicada esta semana en Nature Communications, confirma un nuevo papel de POT1 en relación al desarrollo de diversas formas de cáncer hereditario.
La clave está en los telómeros. Cuando hay anomalías en ellos, los cromosomas se vuelven inestables, lo que a su vez provoca una gran inestabilidad del genoma. POT1 es una pieza fundamental en los telómeros, que funciona mal cuando está mutada.
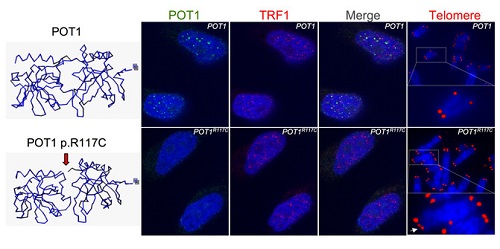
La mutación ahora identificada está en una región de POT1 muy conservada evolutivamente y, por tanto, importante. Los experimentos realizados tanto con modelos estructurales como en células, por Oriol Calvete y Paula Martínez respectivamente, desvelan que debido a esta mutación la proteína que sintetiza POT1 no puede unirse a los telómeros. El resultado es una gran inestabilidad genómica y, en consecuencia, una mayor susceptibilidad al cáncer.
Martínez y Blasco trabajan en el desarrollo de un modelo animal con esa mutación para desarrollar nuevos tratamientos contra tumores humanos con la misma mutación. Otra de las líneas que se abre ahora es tratar de entender el papel de esta mutación en el angiosarcoma cardiaco esporádico, no familiar.
Referencia bibliográfica:
Oriol Calvete, Paula Martinez, Pablo Garcia-Pavia, Carlos Benitez-Buelga, Beatriz Paumard-Hernández, Victoria Fernandez, Fernando Dominguez, Clara Salas, Nuria Romero-Laorden, Jesus Garcia-Donas, Jaime Carrillo, Rosario Perona, Juan Carlos Triviño, Raquel Andrés, Juana María Cano, Bárbara Rivera, Luis Alonso-Pulpon, Fernando Setien, Manel Esteller, Sandra Rodíguez-Perales, Gaelle Bougeard, Tierry Frebourg, Miguel Urioste, Maria A. Blasco, Javier Benítez. A mutation in the POT1 gene is responsible for cardiac angiosarcoma in TP53-negative Li–Fraumeni-like familias. Nature Communications (2015). doi: 10.1038/ncomms9383
El trabajo ha sido financiado por el Gobierno de España, el Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Fondos FEDER, la Unión Europea y la Fundación Botín y Banco Santander a través de Santander Universidades, entre otros.
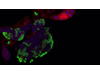
Foto: La sustitución del aminoácido arginina por una cisteina en la posición 117 en POT1p.R117C produce cambios conformacionales en la proteína incompatibles con una unión eficiente al telómero, induciendo un alargamiento telomérico aberrante y telómeros frágiles en las células mutadas. /CNIO

La mejor actitud que podemos adoptar es la de trat...

El equipo de investigadores observó cambios en el...

El gen AtCDF3 promueve una mayor producción de az...

En nuestro post hablamos sobre este interesante tipo de célula del si...

Curapath se complace en reconocer el importante avance logrado por Mac...