Un artículo publicado en la revista Human Genetics por Manel Esteller, Director del Programa de Epigenética y Biología del Cáncer (PEBC) del Instituto de Investigación Biomédica de Bellvitge (IDIBELL), Investigador ICREA y Profesor de Genética de la Universidad de Barcelona, descubre mutaciones en genes asociados con el desarrollo de Síndrome de Rett atípico y distingue esta entidad clínica de otras alteraciones del neurodesarrollo infantil. Este estudio ha sido posible por las ayudas recibidas desde las asociaciones catalana y española del Síndrome de Rett, una campaña de micromecenazgo en Verkami y la Fundación Finestrelles.
"Hemos secuenciado un total de 61 genomas correspondientes a pacientes clasificados como Síndrome de Rett atípico y de sus progenitores, en los cuales no se habia encontrado ninguna alteración en los genes típicamente asociados a esta entidad. Esta tarea compleja de analisis genómico nos ha permitido tener una visión más clara de lo que esta sucediendo a nivel molecular cuando se diagnostica un Síndrome de Rett atípico”, comenta el Dr. Manel Esteller. “A veces ocurre que la mutación responsable se encuentra en alguno de los genes ya conocidos del Síndrome de Rett, pero en regiones inaccesibles a los análisis genéticos más rutinarios. Otras veces la paciente presenta síntomas que parecen un Síndrome de Rett atípico, pero en realidad debido a la similitud del cuadro clínico las mutaciones ocurren en otros genes responsables de síndromes del neurodesarrollo similares como el Síndrome de Dravet, Síndrome de Pitt-Hopkins o enfermedades monogénicas caracterizadas por epìlepsia infantil.”
“Finalmente hemos descubierto otros dos subgrupos muy interesantes - explica el investigador -, aquellas niñas que presentan mutaciones en genes nuevos asociados por primera vez con el Síndrome de Rett atípico, como las encontradas en receptores de neurotransmisores sinápticos; y la existencia de pacientes que, a pesar del análisis detallado de su genoma, no hemos encontrado ninguna mutación evidente siendo posible la existencia de mecanismos alternativos no-genéticos de la enfermedad.”
El Síndrome de Rett es la segunda causa más frecuente de discapacidad intelectual en mujeres por detrás del Síndrome de Down. La principal causa genética del Síndrome de Rett es la aparición de mutaciones en el embrión afectando al gen MECP2, un regulador de la expresión de otros genes del genoma. No obstante, existen pacientes con síntomas muy similares al Síndrome de Rett clásico que no presentan mutaciones en este gen; se trataría de los denominados Síndromes de Rett atípicos. Al desconocerse el gen o genes causantes de los mismos se dificulta su diagnóstico, el consejo genético, su seguimiento y la posibilidad de que puedan beneficiarse de una terapia futura.
Más información:
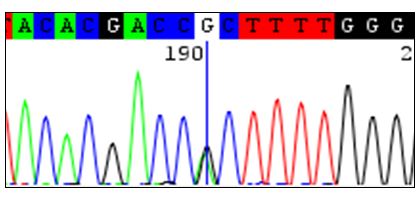
Imagen: Secuenciación del gen de un receptor de un neurotransmisor sináptico en una paciente afectada por Síndrome de Rett atípico. La paciente presenta una mutación en el nucleótido “G” del ADN marcado con una línea azul.

La mejor actitud que podemos adoptar es la de trat...

El equipo de investigadores observó cambios en el...

El gen AtCDF3 promueve una mayor producción de az...

En nuestro post hablamos sobre este interesante tipo de célula del si...

Curapath se complace en reconocer el importante avance logrado por Mac...