Un nuevo artículo publicado en la revista Cell Reports describe como un nuevo fármaco es capaz de disminuir la sintomatología y activar las neuronas aletargadas características del Síndrome de Rett en modelos preclínicos. El estudio, liderado por el Dr. Manel Esteller, Director del Programa de Epigenética y Biología del Cáncer (PEBC) del Instituto de Investigación Biomédica de Bellvitge (IDIBELL), Investigador ICREA y Catedrático de Genética de la Universidad de Barcelona, y la Dra. Sonia Guil, investigadora del mismo grupo del IDIBELL, ha sido posible gracias a las ayudas recibidas desde las asociaciones catalana y española del Síndrome de Rett, la Regata Carla, una campaña de micromecenazgo en Verkami, la Fundación Jérôme Leujene y el proyecto Dischrom de la UE.
El Síndrome de Rett es la segunda causa más frecuente de discapacidad intelectual en mujeres por detrás del Síndrome de Down. La principal causa genética del Síndrome de Rett es la aparición de mutaciones en el embrión afectando al gen MECP2, un regulador de la expresión de otros genes del genoma. No existe un tratamiento farmacológico específico para la enfermedad, por lo que los esfuerzos actuales se centran en intentar controlar las manifestaciones más graves de la misma, como las crisis epilépticas y respiratorias.
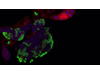
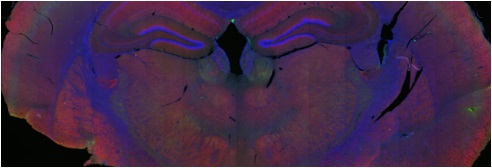
"Sabíamos desde hace unos años que el cerebro de las niñas con Síndrome de Rett se encontraba inflamado; así pues, decidimos probar si un fármaco que inhibe a una proteína central de la neuroinflamación denominada Glicógeno sintasa quinasa-3B (GSK3B) podría revertir parte del cuadro clínico de la enfermedad. Como en todo tratamiento experimental, empezamos con un modelo preclínico de la enfermedad, estudiándolo en ratones que poseen la misma deficiencia MECP2 que en Síndrome de Rett en humanos.” - comenta el Dr. Manel Esteller
“Los resultados han sido muy prometedores, el agente SB216763 ha sido capaz de alargar el tiempo de vida de estos animales enfermos, disminuyendo además notablemente los temblores, las dificultades de respiración y las limitaciones de movilidad. Pero lo verdaderamente destacable es que la inhibición de GSK3B también provoca un “despertar” de las neuronas dormidas del síndrome: estas células cerebrales empiezan ahora a recuperar contactos entre ellas y se incrementa la comunicación entre las sinapsis de las neuronas”, explica el investigador IDIBELL, y concluye: “Nuestros hallazgos proporcionan una nueva vía que puede mejorar la calidad de vida de estas pacientes y ahora es tarea de los neurólogos demostrar su aplicabilidad en las pacientes con Síndrome de Rett. En todo caso, tenemos que ser conscientes de que la mutación en el gen MECP2 sigue ahí, y solo corrigiéndola llegaríamos a un tratamiento definitvo de la enfermedad”.
Más información:

La mejor actitud que podemos adoptar es la de trat...

El equipo de investigadores observó cambios en el...

El gen AtCDF3 promueve una mayor producción de az...

En nuestro post hablamos sobre este interesante tipo de célula del si...

Curapath se complace en reconocer el importante avance logrado por Mac...