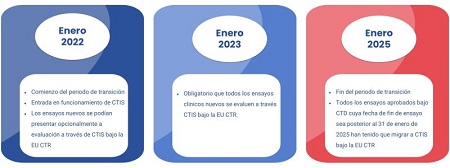
La gestión de los ensayos clínicos sufrió un cambio profundo en enero de 2022, con la entrada en vigor completa del Reglamento (UE) n.º 536/2014 (CTR: Clinical Trials Regulation). Este cambio trajo consigo la creación y el inicio del funcionamiento de CTIS (Clinical Trials Information System), una plataforma centralizada para la gestión, evaluación y supervisión de ensayos clínicos en toda la Unión Europea. Ese año marcó el comienzo del periodo de transición desde la legislación anterior aplicable a los ensayos clínicos, la Directiva 2001/20/CE (CTD), al nuevo Reglamento.
El Reglamento preveía un periodo de tres años, hasta el 31 de enero de 2025, para permitir la migración de todos los ensayos clínicos en curso autorizados bajo la CTD hacia el sistema CTIS. Durante este tiempo, los promotores debían preparar, presentar y obtener aprobación para la transición de sus estudios. Era un periodo complejo para todos los actores, tanto las grandes industrias farmacéuticas, biotecnológicas, como la investigación académica, así como el resto de entidades implicadas, como por ejemplo las CROs.
A enero de 2025, 4.934 ensayos clínicos habían solicitado formalmente la transición al nuevo sistema, según datos recogidos por la EMA, la Comisión Europea y el HMA. Como dato adicional, desde Sermes CRO hemos colaborado en la transición de 117 ensayos clínicos.
En la siguiente imagen se puede ver el numero de solicitudes de transición en los últimos años:
European Comission, European Medicines Agency, Heads of Medicines Agency. Monitoring the European clinical trials environment. A deliverable of the ACT EU Priority Action 2: December 2024. Enero 2025. Accedido en abril 2025. https://accelerating-clinical-trials.europa.eu/document/download/5d300ffd-f2b0-4499-9549-e7b51a5fd851_en?filename=ACT%20EU%20KPI%20Report_December_2024.pdf
Esta situación conlleva importantes consecuencias legales y operativas, ya que los promotores de estos ensayos clínicos podrían enfrentar medidas correctivas y sanciones por parte de los Estados Miembros (EM), conforme a los artículos 77 y 94 del EU CTR, y estar sujetos a responsabilidad civil y penal según el artículo 95 del citado reglamento. Cada Estado Miembro tendrá la potestad de decidir qué medidas aplicar en función del contexto particular de cada estudio afectado.
¿Qué medidas correctivas se pueden implementar en los ensayos que no han realizado a tiempo la transición?
La decisión de aplicar medidas correctivas se evaluará de forma individualizada para cada caso. Si los Estados Miembros deciden implementar estas medidas, se dará al promotor la oportunidad de presentar alegaciones en un plazo de 7 días.
Cuando los Estados Miembro decidan adoptar una medida correctiva bajo el artículo 77 del EU CTR, podrán optar por revocar la aprobación previamente concedida y/o suspender la aprobación. Todo ello busca salvaguardar la seguridad de los participantes y asegurar la integridad de los datos clínicos generados.
¿Qué deben hacer los promotores de los ensayos que no han realizado a tiempo la transición?
Los promotores de ensayos clínicos aprobados por la CTD que sigan en curso y no tengan una aprobación del EU CTR a 31 de enero de 2025, deberán ponerse en contacto con las Autoridades Nacionales Competentes de sus ensayos para iniciar los procedimientos adecuados. En caso de tratarse de un ensayo multinacional, se contactará con el Estado Miembro de Referencia (RMS) que se proponga para la solicitud de autorización bajo el EU CTR.
Por Blanca Oñoro Pedregosa, experta en Regulación Clínica

La mejor actitud que podemos adoptar es la de trat...

The research team observed changes in head circumf...

AtCDF3 gene induced greater production of sugars a...

En nuestro post hablamos sobre este interesante tipo de célula del...

Telum Therapeutics, a biotechnology company leveraging proprietary met...