Las mutaciones del gen MAGEL2, que causan el síndrome de Schaaf-Yan (SYS) —una enfermedad ultrarara que afecta al desarrollo neuronal y cognitivo—, generan proteínas truncadas no funcionales que tienden a acumularse en el núcleo celular. Además, esta acumulación progresiva de proteínas anómalas podría causar un efecto tóxico en los pacientes afectados por el síndrome, que sufren malformaciones congénitas, retraso intelectual, alteraciones en los rasgos faciales, apnea del sueño y contracturas articulares.
Estos avances en la investigación sobre el SYS aparecen en estudio publicado en la revista Journal of Medical Genetics. El estudio lo ha liderado un equipo de la Facultad de Biología y del Instituto de Biomedicina de la Universidad de Barcelona (IBUB), el Instituto de Investigación Sant Joan de Déu (IRSJD) y el Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER). Cabe recordar que este equipo también es autor de la publicación de la primera guía clínica sobre el síndrome de Schaaf-Yang (Journal of Medical Genetics, 2022), dirigida a profesionales sanitarios y familias de niños afectados por esta patología.
Comprender mejor la función, las variantes genéticas y el impacto de la retención nuclear de la proteína MAGEL2 abrirá nuevas vías para diseñar terapias genéticas específicas dirigidas a los pacientes, a fin de evitar la síntesis de la proteína alterada y abordar el SYS, una enfermedad todavía sin tratamiento.
Mutaciones genéticas que originan proteínas truncadas
El gen MAGEL2 se encuentra en el cromosoma 15, se expresa en el sistema nervioso y produce la proteína MAGEL2, implicada en el transporte retrógrado y el reciclaje de proteínas en el citoplasma celular de las neuronas. Hasta ahora, se han documentado en la bibliografía científica más de ochenta mutaciones en el gen MAGEL2, algunas de las cuales se encuentran repetidas entre pacientes. Actualmente, se calcula que hay cerca de 250 personas diagnosticadas con síndrome de Schaaf-Yang en todo el mundo.
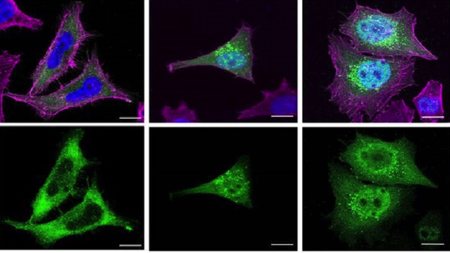
El nuevo trabajo, hecho con células humanas in vitro, constata cómo casi todas las proteínas truncadas asociadas con el síndrome de Schaaf-Yang pierden una parte de la estructura molecular a causa de las mutaciones genéticas. «Las proteínas MAGEL2 funcionales tienen una estructura molecular completa que les permite interactuar con otras proteínas y llevar a cabo sus funciones biológicas normales. Suelen encontrarse en ubicaciones específicas dentro del citoplasma de la célula, principalmente en compartimentos subcelulares relacionados con el transporte y el reciclaje de proteínas», detalla Susanna Balcells, catedrática del Departamento de Genética, Microbiología y Estadística UB, y coordinadora del trabajo. «En cambio —continúa— las proteínas truncadas son versiones más cortas de la proteína MAGEL2, ya que se han visto afectadas por mutaciones genéticas. Por tanto, las proteínas truncadas carecen de ciertas regiones necesarias para funcionar correctamente en la célula».
A causa de las mutaciones genéticas, las proteínas truncadas pierden dominios estructurales clave, como el dominio de homología MAGE, que es crucial para las interacciones con otras proteínas. «La ausencia de este dominio podría impedir estas interacciones esenciales para el correcto funcionamiento de MAGEL2, como su papel en el transporte retrógrado y el reciclaje de proteínas», explica Roser Urreizti.
Cuando las proteínas tóxicas se acumulan en el núcleo de la célula
Las proteínas truncadas suelen acumularse dentro del núcleo celular, y esto podría agravar todavía más la sintomatología de las personas afectadas por el Schaaf-Yang. Como detalla Mónica Centeno, «es probable que, en un contexto celular real, una parte de las proteínas truncadas sintetizadas se puedan encontrar también en ubicaciones específicas dentro del citoplasma, como, por ejemplo, los endosomas. Ahora bien, como su estructura está alterada, tal vez no serían capaces de hacer sus funciones normales correctamente».
La gravedad de las manifestaciones clínicas en los afectados por el síndrome de Schaaf-Yang se podría relacionar con la acumulación de proteínas alteradas en el núcleo celular. «Es decir, todo indica que las mutaciones que provocan síntomas más graves también hacen que la proteína MAGEL2 truncada se acumule más en el núcleo. Esto podría explicarse porque una acumulación superior de proteínas truncadas en el núcleo podría estar interfiriendo con procesos nucleares importantes y afectar en mayor grado el funcionamiento de la célula en condiciones normales», afirma Raquel Rabionet, .
A la búsqueda de tratamientos para abordar el síndrome de Schaaf-Yang
Los mecanismos celulares que impulsan la degradación de proteínas —por ejemplo, el sistema ubiquitina-proteasoma— podrían ayudar a reducir los efectos negativos de las proteínas truncadas y contribuir a mitigar la evolución de la patología. Sin embargo, si la tasa de producción de proteínas supera la capacidad de degradación de la célula, las proteínas aberrantes podrían escapar a la degradación y continuar ejerciendo efectos tóxicos. Tal como explica Aina Prat, «en este sentido, hemos comprobado que las proteínas MAGEL2 normales y truncadas tienen una vida media muy similar. Por tanto, la MAGEL2 truncada se encontraría estable en la célula, donde podría estar ejerciendo los efectos tóxicos».
«Si conociéramos cómo las proteínas MAGEL2 truncadas alteran la función celular, se podrían desarrollar estrategias para promover la degradación de estas proteínas tóxicas, restaurar la función celular o compensar las disfunciones metabólicas y de señalización causadas por su acumulación», concluye el equipo, que impulsará nuevas investigaciones para contribuir al desarrollo de tratamientos innovadores para las personas afectadas por el SYS.
Imagen: Un equipo de la UB, el IBUB, el IRSD y el CIBERER constata cómo las mutaciones en el gen MAGEL2—causante del síndrome— dan lugar a proteínas truncadas y no funcionales que se acumulan en el núcleo celular y, así, podrían agravar los síntomas de los afectados por la patología.
Artículo de referencia: Centeno, Mónica; Alcaide-Consuegra, Estefanía; Gibson, Sophie; Prat-Planas, Aina; Gutiérrez-Ávila, Juan D.; Grinberg, Daniel; Urreizti, Roser; Rabionet, Raquel; Balcells, Susanna. «Subcellular localisation of truncated MAGEL2 proteins: insight into the molecular pathology of Schaaf-Yang syndrome». Journal of Medical Genetics, marzo de 2024. DOI: 10.1136/jmg-2024-109898

La mejor actitud que podemos adoptar es la de trat...

El equipo de investigadores observó cambios en el...

El gen AtCDF3 promueve una mayor producción de az...

En nuestro post hablamos sobre este interesante tipo de célula del si...

Un artículo publicado en Alzheimer’s & Dementia: The Journal of the...










